Analysis pipeline¶
Overview¶
The next generation DNA sequencing based technology utilize RNA proximity ligation to transfrom RNA-RNA interactions into chimeric DNAs. Through sequencing and mapping these chimeric DNAs, it is able to achieve high-throughput mapping of nearly entire interaction networks. RNA linkers were introduced to mark the junction of the ligation and help to split the chimeric RNAs into two interacting RNAs. This bioinformatic pipeline is trying to obtain the strong interactions from raw fastq sequencing data. The major steps are:
- Step 1: Split library based on barcode.txt.
- Step 2: Remove PCR duplicates.
- Step 3: Recover fragments for each library.
- Step 4: Split partners and classify different types of fragments.
- Step 5: Align both parts of “Paired” fragment to the genome.
- Step 6: Annotate aligned RNAs and collapse RNAs mapped to different isoforms.
- Step 7: Link RNA1 and RNA2 to recover interactions
- Step 8: Determine strong interactions.
Pipeline¶
Step 1: Split library based on barcode.txt.¶
Starting from the raw pair-end sequencing data, the libraries from different samples should be separated based on 4nt barcodes in the middle of random indexes (“RRRBBBBRRR”; R: random, B: barcode). It is implemented by split_library_pairend.py
usage: split_library_pairend.py [-h] [-f | -q] [-v] [-b BARCODE]
[-r RANGE [RANGE ...]] [-t] [-m MAX_SCORE]
input1 input2
Example: split_library_pairend.py -q example.R1.fastq
example.R2.fastq -b barcode.txt
positional arguments:
input1 input fastq/fasta file 1 for pairend data (contain
barcodes)
input2 input fastq/fasta file 2 for pairend data
optional arguments:
-h, --help show this help message and exit
-f, --fasta add this option for fasta input file
-q, --fastq add this option for fastq input file
-v, --version show program's version number and exit
-b BARCODE, --barcode BARCODE
barcode file
-r RANGE [RANGE ...], --range RANGE [RANGE ...]
set range for barcode location within reads,default is
full read
-t, --trim trim sequence of 10nt index (do NOT specify this argument if step1 is followed by Step2: Remove PCR duplicates)
-m MAX_SCORE, --max_score MAX_SCORE
max(mismatch+indel) allowed for barcode match,
otherwise move reads into 'unassigned' file
default: 2.
Library dependency: Bio
Here is a example for barcode.txt
ACCT
CCGG
GGCG
The output of this script are several pairs of fastq/fasta files prefixed with the 4nt barcode sequences, together with another pair of fastq/fasta files prefixed with ‘unassigned’.
For example, if the input fastq/fasta files are example.F1.fastq and example.R1.fastq, and the barcode file is the same as above, then the output files are:
- ACCT_example.F1.fastq
- ACCT_example.R1.fastq
- CCGG_example.F1.fastq
- CCGG_example.R1.fastq
- GGCG_example.F1.fastq
- GGCG_example.R1.fastq
- unassigned_example.F1.fastq
- unassigned_example.R1.fastq
Step 2: Remove PCR duplicates.¶
After splitting the libraries, the later steps from here (Step 2-7) need to be executed parallelly for each sample.
With libraries separated, PCR duplicates should be removed from each library if both the 10nt random indexes and the remaining sequences are exactly the same for two pairs. It is achieved by remove_dup_PE.py
usage: remove_dup_PE.py [-h] reads1 reads2
Remove duplicated reads which have same sequences for both forward and reverse
reads. Choose the one appears first.
positional arguments:
reads1 forward input fastq/fasta file
reads2 reverse input fastq/fasta file
optional arguments:
-h, --help show this help message and exit
Library dependency: Bio, itertools
The program will generate two fastq/fasta files after removind PCR duplicates and report how many read pairs has been removed. The output are prefixed with ‘Rm_dupPE’
Note
One pair is considered as a PCR duplicate only when the sequences of both two ends (including the 10nt random index) are the exactly same as any of other pairs.
After that, you may want to trim the 10-nt index off the reads. This can be done by a separate program Remove_index.py
usage: Remove_index.py input_fastq output_fastq
input_fastq: the forward output fastq file of remove_dup_PE.py. For example: Rm_dupPE_ACCT_example.F1.fastq
example: Remove_index.py Rm_dupPE_ACCT_example.F1.fastq Rm_dupPE_ACCT_example.F1_trim.fastq
You may want to rename the reverse output fastq file of remove_dup_PE.py to match the two files.
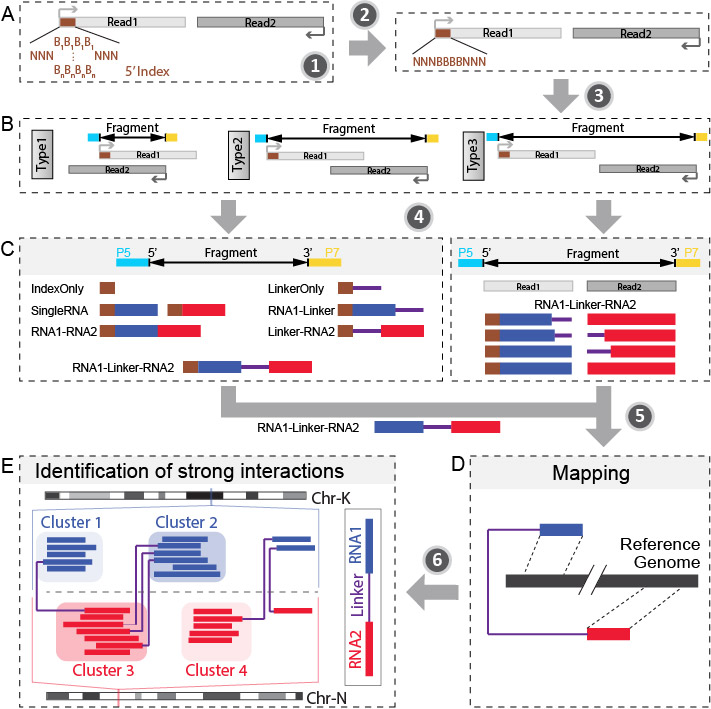
Step 3: Recover fragments for each library.¶
In this step, we are trying to recover the fragments based on local alignment. The fragments are classifed as several different types as shown in the figure below. The flow chart is also clarified at the top.
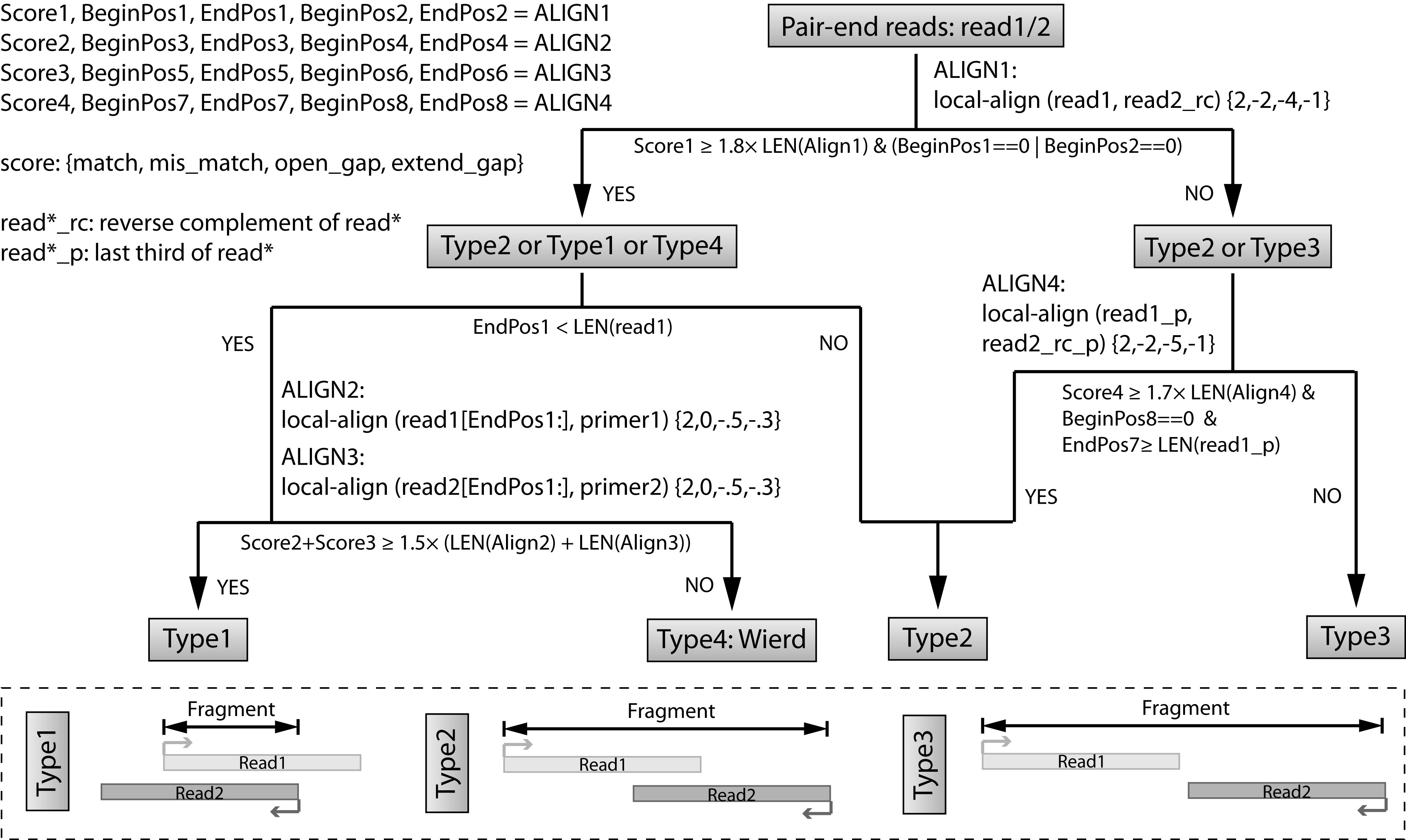
We will use a complied program recoverFragment to do that. Please note that this program supports multi-threading. We highly recommend you to specify the argument -t to increase the speed
recoverFragment - recover fragment into 4 different categories from pair-end seq data
=====================================================================================
SYNOPSIS
DESCRIPTION
-h, --help
Displays this help message.
--version
Display version information
-I, --inputs STR
input of forward and reverse fastq file, path of two files separated by SPACE
-p, --primer STR
fasta file contianing two primer sequences
-t, --threads NUMBER
number of threads to use
-v, --verbose
print alignment information for each alignment
EXAMPLES
recoverFragment -I read_1.fastq read_2.fastq -p primer.fasta -t 8
store fragment using fasta/fastq into 4 output files
'short_*', 'long_*','evenlong_*','wierd_*'
VERSION
recoverFragment version: 0.1
Last update January 2015
Step 4: Split partners and classify different types of fragments.¶
When we recovered the fragments, the next we are goting to do is to find RNA1 and RNA2 that are seprarated by the linkers, and from here, we will be able to classify the fragments into different types: “IndexOnly”, “NoLinker”, “LinkerOnly”, “BackOnly”, “FrontOnly”, “Paired”. (see the figure below).
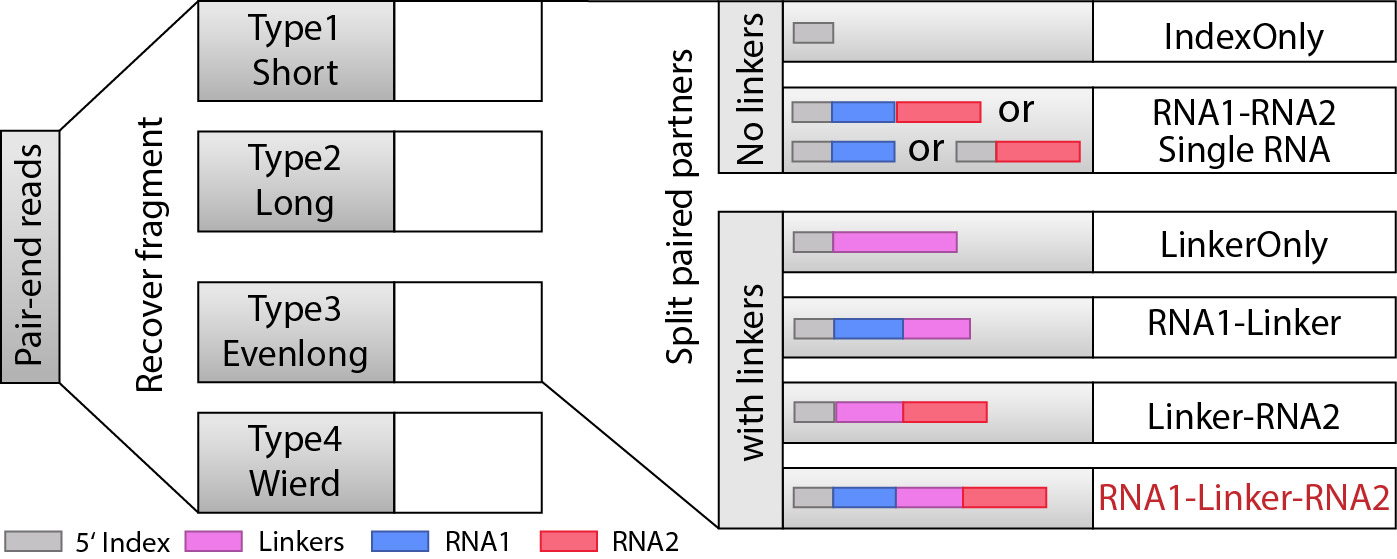
This will be done by split_partner_mp.py. This program also supports multi-processing, and we recommend you to specify the parameter to increase the speed
usage: split_partner_mp.py [-h] [-e EVALUE] [--linker_db LINKER_DB]
[--blast_path BLAST_PATH] [-o OUTPUT] [-t TRIM]
[-b BATCH] [-l LENGTH] [-p PARALLEL]
input type3_1 type3_2
DESCRIPTION: Run BLAST, find linker sequences and split two parts connected by
linkers
positional arguments:
input the input fasta file containing fragment sequences of
type1 and type2
type3_1 read_1 for evenlong (type3) fastq file
type3_2 read_2 for evenlong (type3) fastq file
optional arguments:
-h, --help show this help message and exit
-e EVALUE, --evalue EVALUE
cutoff evalues, only choose alignment with evalue less
than this cutoffs (default: 1e-5).
--linker_db LINKER_DB
BLAST database of linker sequences
--blast_path BLAST_PATH
path for the local blast program
-o OUTPUT, --output OUTPUT
output file containing sequences of two sepatated
parts
-t TRIM, --trim TRIM trim off the first this number of nt as index,
default:10
-b BATCH, --batch BATCH
batch this number of fragments for BLAST at a time.
default: 200000
-r, --release set to allow released criterion for Paired fragment in
Type 3, include those ones with no linker in two reads
-l LENGTH, --length LENGTH
shortest length to be considered for each part of the
pair, default: 15
-p PARALLEL, --parallel PARALLEL
Number of threads
Library dependency: Bio, itertools
Note
The program allows two different strategies for selection of “Paired” fragments from the Type3 fragments. The --release option will allow a read pair to be called as “Paired” fragment even when the linker are not detected in both reads.
The linker fasta file contain sequences of all linkers
>L1
CTAGTAGCCCATGCAATGCGAGGA
>L2
AGGAGCGTAACGTACCCGATGATC
The output fasta files will be the input file name with different prefix (“NoLinker”, “LinkerOnly”, “BackOnly”, “FrontOnly”, “Paired”) for different types. The other output file specified by -o contains information of aligned linker sequences for each Type1/2 fragment.
For example, if the commend is
split_partner_mp.py fragment_ACCT.fasta evenlong_ACCTRm_dupPE_stitch_seq_1.fastq
evenlong_ACCTRm_dupPE_stitch_seq_2.fastq
-o fragment_ACCT_detail.txt --linker_db linker.fa -p 20 --blast_path /usr/bin/blastn
- Then, the output files will be:
- backOnly_fragment_ACCT.fasta
- NoLinker_fragment_ACCT.fasta
- frontOnly_fragment_ACCT.fasta
- Paired1_fragment_ACCT.fasta
- Paired2_fragment_ACCT.fasta
- fragment_ACCT_detail.txt
The format of the last output file fragment_ACCT_detail.txt will be “Name | linker_num | linker_loc | Type | linker_order”. Here are two examples:
HWI-ST1001:238:H0NYEADXX:1:1101:10221:1918 L1:2;L2:1 19,41;42,67;68,97 None L2;L1;L1
HWI-ST1001:238:H0NYEADXX:1:1101:4620:2609 L1:2 28,46;47,79 Paired L1;L1
In the first fragment, there are three regions can be aligned to linkers, 2 for L1 and 1 for L2, the order is L2, L1, L1. And they are aligned in region [19,41], [42,67], [68,97] of the fragment. “None” means this fragment is either ‘LinkerOnly’ or ‘IndexOnly’ (in this case it is ‘LinkerOnly’). This fragment won’t be written to any of the output fasta files.
In the second fragment, two regions can be aligned to linkers, and they are both aligned to L1. The two regions are in [28,46], [47,79] of the fragment. the fragment is “Paired” because on both two sides flanking the linker aligned regions, the length is larger than 15nt. The left part will be writen in Paired1_fragment_ACCT.fasta and the right part in Paired2_fragment_ACCT.fasta
For some experimental designs random nucleotides will be added to one or two ends of the linker (“NNlinkerNN”; N:random nucleotide). You can use the program Remove_random.py to trim them after splitting the two reads
usage: Remove_random.py [-h] [-H HEAD] [-t TAIL] input output
Remove random nucleotides from the head and/or tail of reads
positional arguments:
input: Input fasta file
output: output fasta file
optional arguments:
-h, --help show this help message and exit
-H HEAD, --head HEAD Number of nucleotides cut at head. default: 0
-t TAIL, --tail TAIL Number of nucleotides cut at tail. default: 0
Step 5: Align both parts of “Paired” fragment to the genome.¶
In this step, we will use the Paired1* and Paired2* fasta files output from the previous step. The sequences of part1 and part2 are aligned to the specified genome/transcriptome/microRNA database with Bowtie2/blast. This is implemented using script Stitch-seq_Aligner.py.
usage: Stitch-seq_Aligner.py [-h] [-bt bowtie_path | -bt2 bowtie2_path]
[-bl blat_path] [-f fastq_to_fasta_path]
[-s samtool_path] [--ref [rr [rr ...]]]
[--reftype [rt [rt ...]]] [-P PARTNUM] [-nostr]
[-p NTHREADS] [-r] [-l MIRNALEN] [-o OUTPUT_PATH]
part_reads
Align RNA pairs for Stitch-seq.
positional arguments:
-h, --help show this help message and exit
-bt bowtie_path, --bowtie_path bowtie_path
path for the bowtie executive
-bt2 bowtie2_path, --bowtie2_path bowtie2_path
path for bowtie2 executive
-th tophat_path, --tophat_path tophat_path
path for tophat executive
-bl blat_path, --blat_path blat_path
path for the blat executive (for miRNA only)
-f fastq_to_fasta_path, --fastq_to_fasta_path fastq_to_fasta_path
path for the fastq_to_fasta executive (for miRNA only)
-s samtool_path, --samtools_path samtool_path
path for samtools executive
--ref [rr [rr ...]] Reference seq files, references will be used in the
order specified
--reftype [rt [rt ...]]
Reference types: (miRNA, genome, transcript, other)
-P PARTNUM, --part_num PARTNUM
Specify which part (1 or 2) of the paired read is
being mapped (this is used for strand matching
purposes).
-nostr, --ignore_strand
Reads mapped onto the wrong strand will be considered
as not mapped by default. Set this flag to ignore
strand information.
-p NTHREADS, --threads NTHREADS
Number of threads used in bowtie mapping.
-r, --resume Set to let Stitch-seq recover from previous files.
Parameters other than number of threads need to be
exactly the same for recovery. This may be useful if
Stitch-seq crashes for CPU/memory/storage reasons.
-l MIRNALEN, --mirnalen MIRNALEN
Set the maximum length allow for a miRNA alignment
(default = 35), a higher value may recover more miRNA
alignments if there are references at such length but
will be slower.
-o OUTPUT_PATH, --output_path OUTPUT_PATH
The path where all output files will be written to.
Input files with the same name may need different
output path values to prevent overwriting. default
value: 'output'
Library dependency: Bio, pysam, itertools
Here is an example using Bowtie2:
Stitch-seq_Aligner.py Paired1_fragment_ACCT.fasta -bt2 /usr/bin/bowtie2 -bl /usr/bin/blat --ref mm9_miRNA.fa mm9_transcripts.fa mm9
--reftype miRNA transcript genome -s samtools -p 8 -P 1 -nostr
If you want to use Tophat, you can refer to the following example:
Stitch-seq_Aligner.py Paired1_fragment_ACCT.fasta -th /usr/bin/tophat -bl /usr/bin/blat --ref mm9_miRNA.fa mm9
--reftype miRNA genome -s samtools -p 8 -P 1 -nostr
The program allows user to map the data on up to three different references: microRNA, genome and transcriptome. References will be used in the order specified. Only reads cannot be mapped in the previous step will be mapped to the next reference. In other words, each read can be mapped to only one reference. Therefore you may want to specify the order of your references. For example, if you have a massive number of mature microRNAs in your sample, you may want to map the data to miRNA reference first, and then to the genome.
Pre-built bowtie2 indexes, transcriptome reference(Ensembl) and microRNA reference(miRbase) for mm9 and hg19 are included in Data/Ref/folder. If you would like to prepare your own reference please see the requirements below:
Transcript reference requirements. Transcript references should be downloaded from Ensembl (preferably with BioMart) with the following header information separated by ‘|’:
| [1] | Or other ID |
| [2] | Or other name annotation |
microRNA reference. MicroRNA references can be downloaded from miRbase. Nucleotides “U” in the fasta file should be replaced by “T”.
The format for the output file ACCT_fragment_paired_align.txt will be:
Column [3] Description 1 chromosome name of RNA1 2,3 start/end position of RNA1 4 strand information of RNA1 5 sequence of RNA1 6 The reference type RNA1 is mapped to 7 RNA type for RNA1 8 RNA name for RNA1 9 RNA subtype [4] for RNA1 10 Whether strand agrees to reference [5] 11 name of the pair 22 [6] ‘multimap’ if the pair has multiple hits
| [3] | column 12-21 are the same as column 1-10 except they are for RNA2 instead of RNA1. |
| [4] | subtype can be intron/exon/utr5/utr3 for lincRNA and mRNA (protein-coding), ‘.’ for others |
| [5] | ‘ProperStrand’ if the strand of the read agrees to the reference, ‘NonProperStrand’ if not. |
| [6] | if there is only one hit for the row or –unique is specified, there will be no 22nd column for this row. |
Note
The program will not filter reads with MAPQ lower than a specific value out. This can be done by step 6 or manually.
Step 6: Annotate aligned RNAs and collapse RNAs mapped to different isoforms.¶
After alignment, we are going to annotate RNAs to identify RNA types and names. The annotation will be done with Stitch-seq_Annotation.py.
usage: Stitch-seq_Annotation.py [-h] [--annotype [at [at ...]]]
[--tx_anno txa] [-a ANNOTATION] [-A DB_DETAIL]
[-R DB_REPEAT] [-M MAPQ] [-P PAIR]
input_path file_perfix
Annotate aligned RNAs.
positional arguments:
bam_files bam file/blatresult that need to be annotated. The
format of input files should be .bam/.blatresult
optional arguments:
-h, --help show this help message and exit
--annotype [at [at ...]]
Reference types: (miRNA, genome, transcript, other).
The order should be the same as that of the
bam/blatresult files
--tx_anno txa Annotation file for transcriptome. It is the reference
for alignment, that is, rna.fa. Should be specified if
annotype contains transcript
-a ANNOTATION, --annotation ANNOTATION
If specified, include the RNA type annotation for each
aligned pair, need to give bed annotation RNA file.
Should be specified if annotype contains genome
-A DB_DETAIL, --annotationGenebed DB_DETAIL
annotation bed12 file for lincRNA and mRNA with intron
and exon. Should be specified if annotype contains
genome
-R DB_REPEAT, --annotationRepeat DB_REPEAT
annotation bed6 file from repeatMasker. Should be
specificed if annotype contains genome
-M MAPQ, --mapq MAPQ For reads mapped to genome, reads with MAPQ lower than
M will be labeled as 'Many' and will be discarded in
next step
-P PAIR, --pair PAIR Specify if the RNA is RNA1/2. Should be 1 or 2
Here is an example
Stitch-seq_Annotation.py output Paired1_fragment_ACCT -P 1 --annotype genome transcript -a mm9_geneAnno.bed -A mm9_geneDetail.bed
-R mm9_repeatMasker.bed --tx_anno mm9_rna.fa -M 2
Annotation files for different types of RNAs in mm9 and hg19 genome were included in Data/Ref/folder.
The output files will be a file suffixed with ”.pairOutput”. For example, if the barcode is ACCT, then the output files will be (depends on which part was the input):
- Paired1_fragment_ACCT.pairOutput
- Paired2_fragment_ACCT.pairOutput
- The format of the .pairOutput file will be:
Column Description 1 chromosome name of RNA1/2 2,3 start/end position of RNA1/2 4 strand information of RNA1/2 5 sequence of RNA1/2 6 the reference type RNA1/2 is mapped to 7 RNA type for RNA1 8 RNA name for RNA1 9 RNA subtype [7] for RNA1 10 whether strand agrees to reference [8] 11 name of the pair 12 unique map or not [9]
| [7] | (1, 2) subtype can be intron/exon/utr5/utr3 for lincRNA and mRNA (protein-coding), ‘.’ for others |
| [8] | (1, 2) ‘ProperStrand’ if the strand of the read agrees to the reference, ‘NonProperStrand’ if not. |
| [9] | For reads mapped to genome, “unique” refers to the MAPQ of the read passed the specific threshold. For reads mapped to transcriptome, this column is useless as a read can be mapped to different isoforms of the same gene. Those lines will be collapsed later. |
For reads mapped to transcriptome, column 1-3 in .pairOutput files will be Ensembl ID of the transcript and start/end position of the reads on the transcript. Therefore, the chromosome and genomic coordinates need to be recovered. Moreover, a read mapped to transcriptome can be mapped to different isoforms of the same gene. All of these results will be output, resulting in dozens of lines for one read in .pairOutput files. These lines can be collapes together for downstream analysis. All of these goals can be achieved with Merging_Transcript.py.
An additional function of this program is to label “dead microRNAs”, that is, microRNAs that have been shown not to be a valid microRNA and removed from miRbase. This can be done with a customer-provided “dead miRNA list”.
Similar to Stitch-seq_Annotation.py, this program only take one input file each time (either RNA1 or RNA2). In other words, one needs to run it twice to merge all of the reads.
usage: Merging_Transcript.py [-h] -i INPUT [-md MIRNA_DEAD] -T
ANNOTATION_TRANS [-o OUTPUT]
Merge fragments mapped to different transcripts of the same gene
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
paired fragments. MUST BE SORTED BY LINKER NAME
-md MIRNA_DEAD, --miRNA_dead MIRNA_DEAD
-T ANNOTATION_TRANS, --annotation_trans ANNOTATION_TRANS
Ensembl annotation file for transcript
-o OUTPUT, --Output OUTPUT
Output file name
The output file will be a file suffixed with ”.pairOutput.merged”. The format of ”.pairOutput.merged” files is exactly the same as that of ”.pairOutput” files. Now column 12 reflects whether or not each read is “uniquely” mapped correctly.
Step 7: Link RNA1 and RNA2 to recover interactions¶
In this step, we will use the two ”.pairOutput.merged” files to link RNA1 and RNA2 together. Only fragments with both parts “uniquely” mapped will be output. We will link the two RNAs with Link_frags.py
usage: Link_frags.py [-h] --RNA1 RNA1 --RNA2 RNA2 [-o OUTPUT]
Link fragment1 and 2 by linker ID. Fragment will be output only when both RNA1
and 2 are uniquely mapped.
optional arguments:
-h, --help show this help message and exit
--RNA1 RNA1 merged RNA1
--RNA2 RNA2 merged RNA2
-o OUTPUT, --output OUTPUT
Output file name.
- The output file `ACCT_RNA_noLeftRight.txt ` will be:
Column[#f10]_ Description 1 chromosome name of RNA1 2,3 start/end position of RNA1 4 strand information of RNA1 5 sequence of RNA1 6 the reference type RNA1 is mapped to 7 RNA type for RNA1 8 RNA name for RNA1 9 RNA subtype [7] for RNA1 10 whether strand agrees to reference [8] 11 name of the pair
| [10] | Column 12-21 will be the same as 1-10 for RNA2 |
Step 8: Determine strong interactions.¶
In this step, we will first count reads mapped to every RNA. Then based on the pairing information, we count the interactions between RNA1 and RNA2. For each interaction, a p-value can be generated based on hypergeometric distribution. Given the p-values of all interactions, we could adjust the p-values controlled by False Discovery Rate (FDR, Benjamini-Hochberg procedure). The strong interactions can be selected by applying a FDR cutoff from adjusted p-values.
We will use the script Select_strongInteraction_RNA_noLeftRight.py to count reads mapped to different RNA-RNA interactions:
usage: Select_strongInteraction_RNA_noLeftRight.py [-h] -i INPUT
[-M MIN_CLUSTERS]
[-m MIN_INTERACTION]
[-p P_VALUE] [-o OUTPUT]
[-O OUTPUT_INTRA]
[-a ANNOTATION]
[-r ANNOTATION_REPEAT]
find strong interactions from paired data
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
input file which is the output file of Stitch-seq-
Aligner.py
-M MIN_CLUSTERS, --min_clusterS MIN_CLUSTERS
minimum number of segments allowed in each cluster,
default:5
-m MIN_INTERACTION, --min_interaction MIN_INTERACTION
minimum number of interactions to support a strong
interaction, default:3
-p P_VALUE, --p_value P_VALUE
the p-value based on hypergeometric distribution to
call strong interactions, default: 0.05
-o OUTPUT, --output OUTPUT
specify output file
-O OUTPUT_INTRA, --output_intra OUTPUT_INTRA
specify output file for intra interactions
-a ANNOTATION, --annotation ANNOTATION
Annotation file for all RNA components
-r ANNOTATION_REPEAT, --annotation_repeat ANNOTATION_REPEAT
Annotation file for all repeat components
need Scipy for hypergeometric distribution
The input of the script is the output of Step 5 (ACCT_fragment_paired_align.txt in the example). “annotated_bed” class is utilized in this script.
Here is a example:
Select_strongInteraction_RNA_noLeftRight.py -i ACCT_linked_fragment.txt -a mm9_geneIDAnno.bed -r mm9_repeatMasker.bed -o ACCT_RNA_noLeftRight.txt -O ACCT_self.txt
The column description for output file ACCT_RNA_noLeftRight.txt is:
Column Description 1 chromosome name of RNA1 2,3 start/end position of cluster in RNA1 4 RNA type of RNA1 5 RNA name of RNA1 6 RNA subtype of RNA1 7 RNA Ensembl ID of RNA1 8 whether strand agrees to reference 9 # of counts for cluster in RNA1 10-18 Same as 1-9, but for RNA2 19 # of interactions between these two clusters 20 log(p-value) of the hypergeometric testing
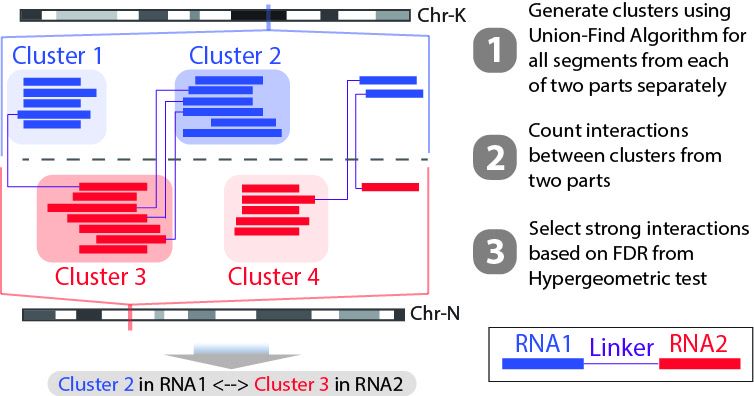
We also have another script Select_strongInteraction_pp.py to determine strong interactions based on interaction sites. Instead of calling strong interactions based on RNA annotations, we will generate clusters with high coverage separately for all RNA1 (R1) and RNA2 (R2) segments. Then based on the pairing information, we count the interactions between clusters from RNA1 and RNA2 (See the figure below). Results of this program are mainly used for data visualization.