MARIO-tools 0.4 documentation¶
Overview¶
MARIO-tools is a set of bioinformatic tools for analysis of a novel DNA sequencing based technology to detect RNA-RNA interactome and RNA-chromatin interactome (RNA-chromatin interactome is coming soon).
MARIO-tools automated all the analysis steps, including removing PCR duplicates, splitting multiplexed samples, identifying the linker sequence, splitting junction reads, calling interacting RNAs, statistical assessments, categorizing RNA interaction types, calling interacting sites, and RNA structure analysis, as well as visualization tools for the RNA interactome (Visualization of global interactome) and the proximal sites within an RNA (Heatmap for Intra-RNA interactions).
Below is a illustration for the experimental design of this new technology. This procedure crosslinks RNAs with their bound proteins, and ligates the RNAs co-bound by the same protein into a chimeric RNA. The chimeric RNA is interspersed by a predesigned biotinylated RNA linker, in the form of RNA1-Linker-RNA2. These linker-containing chimeric RNAs are selected by streptavidin and then subjected to pair-end sequencing
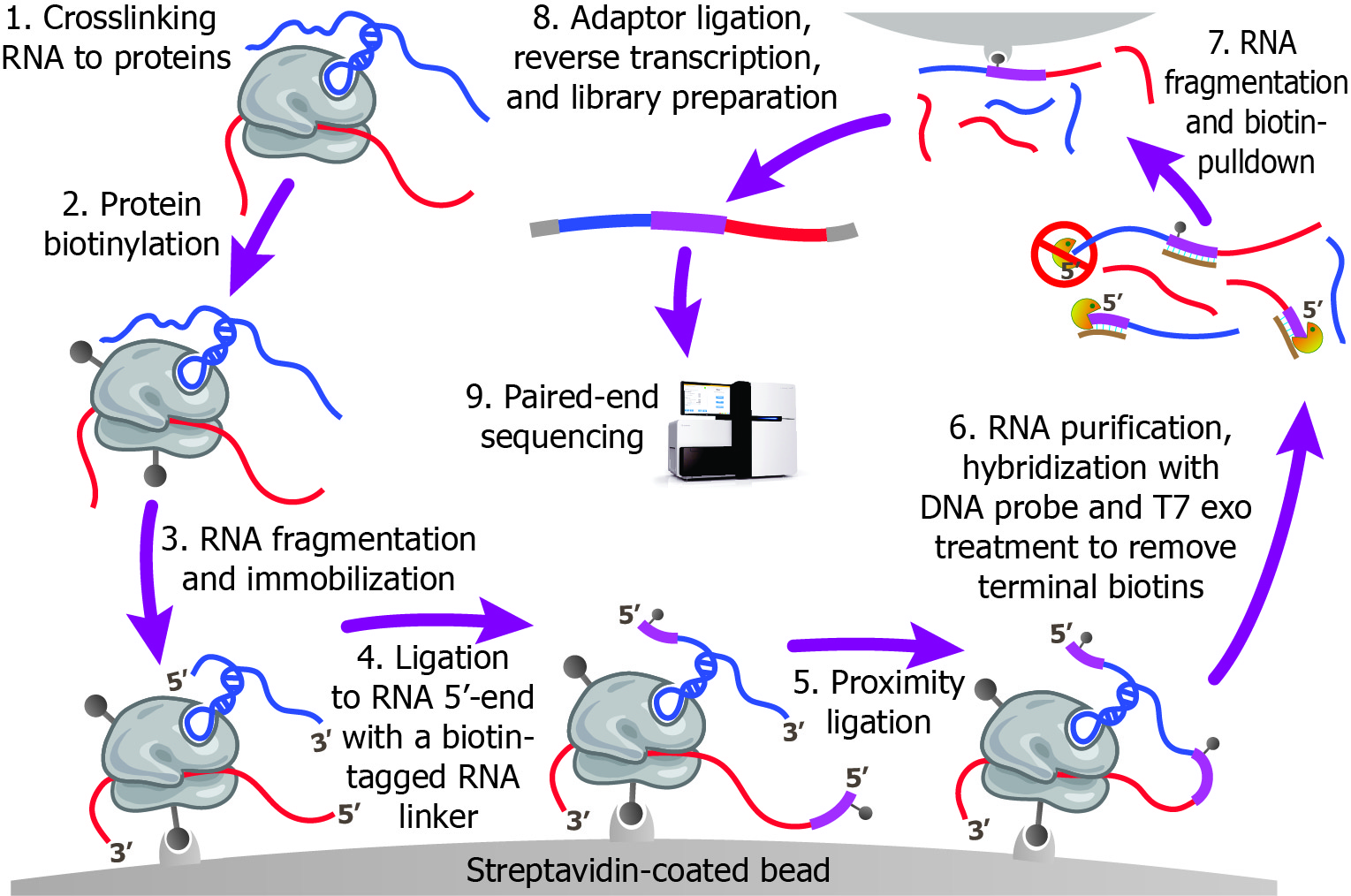
The MARIO method offers several advantages for mapping RNA-RNA interactions. First, the one-to-one pairing of interacting RNAs is experimentally captured. Second, by using the biotinylated linker as a selection marker, it circumvents the requirement for either a protein-specific antibody or expressing a tagged protein, allowing for an as unbiased mapping of the entire RNA interactome as possible. Third, false positive interactions, produced by ligation of random RNAs that happened to be proximal in space, are minimized by performing RNA ligation on streptavidin beads in a dilute condition. Fourth, the predesigned RNA linker provides a clear boundary to split any sequencing read that spans across the ligation spot, thus avoids ambiguities in mapping the sequencing reads. Fifth, MARIO directly analyzes the endogenous cellular condition without introducing any exogenous nucleotides or protein-coding genes before crosslinking. Sixth, potential PCR amplification biases were removed by attaching a random 6nt barcode to each chimeric RNA before PCR amplification, where the completely overlapping sequencing reads with identical barcodes are counted only once.
Installation¶
step 1: Install the dependent prerequisites:¶
- Python libraries [for python 2.x]:
- The Boost.Python C++ library
- Other softwares needed:
- Bowtie (or Bowtie 2 if you set Bowtie2 option in Stitch-seq_Aligner.py)
- Tophat (if you set Tophat option in Stitch-seq_Aligner.py)
- samtools
- NCBI blast+ (use blastn)
Step 2: Download the package¶
Clone the package from GitHub:
git clone https://github.com/Jia340/MARIO.git
Step 3: Add library source to your python path¶
Add these lines into your ~/.bash_profile or ~/.profile
Location="/path/of/MARIO" # change accordingly
export PYTHONPATH="$Location/src:$PYTHONPATH"
export PATH="$PATH:$Location/bin"
Loc_lib="/path/of/boost_1_xx_0/lib/" # change accordingly
export LD_LIBRARY_PATH="$Loc_lib:$LD_LIBRARY_PATH"
Support¶
For issues related to the use of MARIO, or if you want to report a bug or request a feature, please contact Xiaoyi Cao <x9cao at eng dot ucsd dot edu> or Jia Lu <jil340 at eng dot ucsd dot edu>